|
У 40-х роках двадцятого століття вченими Джеймсом Мартіном з Ірландії і англійкою Джулією Белл вперше описана клінічна картина захворювання. Генетики протягом певного часу спостерігали родину, де у абсолютно нормальної жінки народжувалися розумово відсталі хлопчики. Відстеживши фамільну історію, виявили прецеденти у чоловіків в попередніх поколіннях. Вчені встановили причину генетичної аномалії, яка полягала в ламкості дистального плеча X-хромосоми. Візуально зазначалося звуження решт за рахунок вторинної перетяжки на стоншеному ділянці в локусі Xq27-28 (фото). У 90-х роках шляхом цитогенетичного обстеження було виявлено мутований ген, що послужив причиною синдрому фрагільної хромосоми. У назву патології увійшли імена перших учених, що звернули увагу на зміну цього типу в геномі. Захворювання, що передається у спадок, зчеплене з підлогою. У хлопчиків симптоматика явно виражена, у дівчаток зустрічається значно рідше, протікає в легкій формі розумової відсталості. Відноситься до поширеної аномалії (1: 4000), за частотою виникнення займає провідне місце серед спадкових патологій.
Причини виникненняГенотип людини становлять 46 хромосом, дві з них визначають стать - X, Y. У жінок набір 46 XX, у чоловіків - 46 XY. Цим пояснюється рідкісне прояв хвороби у дівчаток, коли відбувається компенсація з боку другої хромосоми каріотипу. Ланцюжок генетичної пам'яті складається з повторюваних комбінацій цитозин-гуанін-гуанін (ЦГГ), збільшення копій (експансія) истончает ділянки ДНК, викликаючи Синдром Мартіна-Белла (СМБ). Аномалія виникає на тлі мутації FMR1, гена, що відповідає за кодування білка, основного учасника формування нервової системи. Відрізки X хромосоми характеризуються чотирма категоріями | стан | Показник чергування трінуклеотідамі | розвиток хвороби |
|---|
| нормальне | 29–30
| відсутність СМБ |
| проміжне (сіра зона) | 44–55
| ризик розвитку |
| премутація | 60–200
| синдром не розвивається, людина є носієм зламаного гена, хвороба проявиться в наступних поколіннях |
| повне порушення ланцюжка | 250–4000
| поява аномалії |
Мутація гена пригнічує функціональність білка, що бере участь в розвитку дитини, впливає на його здатність до навчання і запам'ятовування нового матеріалу. Недостатність ферменту позначається на формуванні аксонів, синапсів, які беруть безпосередню участь в нервових зв'язках, на цьому тлі розвиваються неврологічні відхилення і розумова відсталість.
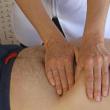
Успадковується генетична патологія по жіночій лінії. У чоловіка, що має одну X-хромосому, після передачі зламаного гена від матері дебют синдрому припадає на дванадцятий місяць життя з подальшою прогресією. Передати аномалію він може тільки дочки. У дівчинки дефіцит поповнюється за рахунок другої хромосоми. Патологія не проявляється, в гіршому випадку супроводжується легкої симптоматикою. Жінка передає синдром ламкої Х-хромосоми потомству обох статей, коло замикається. Тому в межах одного родового клану у чоловіків відзначається порушення розумового розвитку, при цьому жіноча половина абсолютно здорова або з незначними відхиленнями. Характерні ознаки патологіїГенетичне захворювання супроводжується різноманітністю ознак, кожен випадок індивідуальний набором власної симптоматики. СМБ відрізняє від інших неврологічних патологій ряд особливостей: розлад психоемоційного сприйняття, прогресуюча розумова відсталість, відхилення у фізичному розвитку. Синдром Мартіна-Белла у дітей характеризується симптомами, що дозволяють без праці визначити форму генетичної мутації. Новонароджений хлопчик відрізняється великою вагою і збільшеним розміром яєчок (макроорхізм), без гормональних відхилень. Відзначається зниження смоктального і хапального рефлексу, м'язового тонусу, він погано реагує на зовнішні подразники. Дитина відстає від однолітків у фізичному та інтелектуальному розвитку. У більшості випадків з'являється на світ з вродженими захворюваннями: пороками серця, деформацією суглобів. Тому діти з СМБ пізно починають ходити, практично не повзають, мовна функція носить загальмований характер, словниковий запас убогий, дикція невиразна, в важких випадках проявляється повна відсутність комунікативних здібностей, дитина мовчить. У міру прогресії синдрому Мартіна-Бела ознаки доповнюються, мають більш виражений характер. При затримці психомоторного розвитку руху набувають гіперкінетичні особливості: - безсистемне розмахування руками, хлопки в долоні, струшування фалангами;
- стрибки на місці;
- кругові рухи тулубом, повороти навколо своєї осі;
- дискоординація, безглуздість і помпезність поз.
 Психологічні відхилення: - емоційна лабільність (реакція на подразник не відповідає віку);
- безконтрольне прояв гніву, агресії;
- необгрунтоване упертість, плаксивість, дефіцит уваги;
- боязнь фізичного контакту, скупчення малознайомих людей, гучних звуків;
- симптоми аутизму.
Неврологічні аномалії, властиві синдрому: - епілептичні напади на тлі м'язових судом, тимчасової втрати свідомості;
- нервовий тик, локалізований в нижній частині обличчя і століттях, що викликає викривлення міміки і часте моргання;
- тремор верхніх кінцівок;
- гіперподвіжность, хворий нездатний перебувати на одному місці тривалий час:
- окорухові, пірамідні розлади.
Синдром фрагільної хромосоми формує фізичні відхилення в розвитку, хворі хлопчики візуально відрізняються від здорових однолітків: - голова великого розміру з опуклим високим чолом, від цього обличчя набуває форму овалу;
- піднебінний звід глибокий, нижня щелепа важка;
- вуха круглі, відстовбурчені, з низькою посадкою на черепі;
- ніс загострений, у вигляді гачка;
- очі широко поставлені, косять.
Фенотип доповнюється еластичністю шкіри, плоскостопістю, кривизною ніг, широкими ступнями і кистями. У всіх носіїв зламаного FMR1 незмінною симптоматикою є дисфункція щитовидної залози, надниркових залоз. Ендокринні порушення викликають недостатність метаболізму (ожиріння), раннє статеве дозрівання. Рівень розумової відсталості коливається від легкої форми до важкого клінічного перебігу. Основний відсоток пацієнтів знаходиться на стадії олігофренії. Жінки відрізняються високим лібідо, але у них настає ранній період менопаузи. Відбувається переродження яєчників в кістозні новоутворення. У чоловіків відзначається явний макроорхізм. діагностичні дослідженняВизначення синдрому Мартіна-Бела передбачає застосування специфічних тестів для аналізу стану Х-хромосоми на Xq27-28 ділянці. Проводиться лікарем генетиком за наступним алгоритмом: - Огляд пацієнта з урахуванням специфічних змін зовнішності і гипотонуса м'язової маси.
- Основною методикою в діагностиці патології, що дає 100% результат на ранньому етапі клінічного розвитку, є цитогенетичний спосіб. Беруться клітини хворого, обробляються фолієвою кислотою, яка запускає процес зміни хромосоми. Якщо аномалія відзначилася на Xq27-28 лакмус, наявність синдрому не викликає сумнівів.
- На більш пізніх термінах застосовується дослідження пари хромосом, що відповідають за підлогу (каріотипування). Мутація підтверджує СМБ.
- За допомогою поліланцюгової реакції аналізується склад і структуру трінуклеотідамі.
- Молекулярно-генетичне вивчення визначає частоту повторів ЦГГ.
- При синдромі ламкою X-хромосоми у всіх пацієнтів однакова біоелектрична активність головного мозку, що дозволяє за допомогою електроенцефалографії підтвердити діагноз.
Хворобу можна виявити на ранніх стадіях вагітності. Перинатальне обстеження грунтується на УЗД, аналізі сироватки крові жінки, біопсії хоріона. Якщо підтвердилася генетична аномалія у плода, пропонується переривання вагітності, але в будь-якому випадку вибір залишається за майбутньою матір'ю.
 ефективне лікуванняЯк і від будь-якого генетичного захворювання, що передається у спадок, від синдрому Мартіна-Бела позбутися неможливо. Терапія медикаментами проводиться комплексно з фізіопроцедурами, в крайніх випадках вдаються до оперативного втручання. Заходи спрямовані на зменшення симптоматики і покликані поліпшити якість життя, запобігти прогресуванню розумової відсталості і неврологічних відхилень. консервативні методиЛікування генетичної аномалії передбачає застосування таких препаратів: - розріджують кров - «Клексан», «Плавикс»;
- попереджувальних епілептичні напади - «Мазепин»;
- ноотропного дії - «Пірацетам»;
- для поліпшення стану судин і мозкового кровообігу - «Церебролізин», Винпоцетин »;
- седативного (заспокійливого) ефекту - «Седуксен», «Діазепам»;
- антидепресантів - «Сертралін», «Флуоксетин», «Кломіпрамін»;
- соматичного дії (психостимулятори) - «Солкосерил», «Кавінтон», «Лидаза»;
- нейролептиків - «Хлорпромазин», «Галоперидол», «Періціазін».
У комплексній терапії застосовують препарати на основі літію разом з набором вітамінів, які нормалізують когнітивну функцію. Прагнення лікувати синдром фолієвою кислотою виявилося неефективним. Терапія на час покращувала поведінкові і комунікативні можливості, але не гальмувала процес розумової деградації.
фізіотерапіяНа допомогу консервативному впливу на прояви синдрому призначається ряд фізіотерапевтичних заходів: - лікувальна фізкультура;
- вправи в басейні;
- душ Шарко;
- грязьові ванни з радоном;
- акупунктура (голковколювання);
- гірудотерапія (п'явки для розрідження крові);
- м'язова релаксація.
Показані заняття з логопедом, тренінги з психотерапевтом.
оперативне лікуванняХірургічне втручання доцільно, якщо ускладнення синдрому Мартіна-Бела торкнулося життєво важливі органи. Робиться операція при вроджених вадах серця, кістозному переродження яєчників з ризиком переходу в злоякісне утворення. Застосовується пластична корекція, мета якої - усунення фізичних дефектів, властивих хвороби. Хірургічним методом наводяться в норму кінцівки, змінюється форма вух, усувається зовнішня аномалія статевих органів. Прогноз і профілактикаГенетична ламкість X-хромосоми не створює великих проблем зі здоров'ям, а то й ускладнена патологіями внутрішньоутробного розвитку. Тривалість життя не відрізняється від здорових людей. Прогноз на одужання несприятливий. При адекватному симптоматичного лікування, психологічної корекції, допомоги людині з адаптацією в соціумі якість життя значно покращиться, але в підсумку синдром призведе до інвалідності. Профілактикою захворювання є перинатальне обстеження плода на ранніх термінах. Скринінг біологічного матеріалу допоможе виявити мутацію на Xq27-28 ділянці. В цьому випадку рекомендується переривання вагітності. Чоловікові або жінці, в роду яких були прецеденти мутації FMR1, перед плануванням дитини необхідно пройти тест. Якщо аномалія у одного з батьків підтверджується, на молекулярному рівні існують способи виправити дефект X-хромосоми і провести екстракорпоральне запліднення. ЕКО дасть можливість народження немовляти із здоровим генетичним кодом.
Синдром тендітної Х-хромосоми (fragile X syndrome) Маловідома до 1980-х рр., Тендітна Х-хромосома являє собою хромосомну аномалію, яку вважають зараз найбільш частою спадкової причиною затримки розумового розвитку. Синдром Дауна - осн. генетично обумовлена \u200b\u200bпричина розумового недорозвинення - виникає внаслідок невдалого роз'єднання 21-й хромосомної пари при мейозі; він яв-ся генетичним за своєю природою, але не успадкованими. Розміщена на слабкому або крихкому ділянці на Х-хромосомі, fra (X) пов'язана з іолом і тому частіше зустрічається у чоловіків. Це - лише одне з більш ніж 50 пов'язаних з Х-хромосомою розладів, що викликають затримку псіхіч. розвитку, до-рої має велику поширеність. Хоча в різних роботах наводяться різні оцінки, морбідность становить близько 1: 1500 у чоловіків і 1: 2000 у жінок. Fra (X) яв-ся причиною 2-7% випадків розумової відсталості у чоловіків. Вона має дві незвичайні генетичні характеристики: а) гетерозиготні (носії) жінки з однієї нормальної Х-хромосомою і однієї з крихким ділянкою можуть демонструвати зниження інтелекту і недостатню специфічну здатність до навчання; б) близько 20% чоловіків, що успадкували крихкий ділянку, яв-ся непенетрантнимі, не виявляючи помітних фіз. або психол. ефектів і ознак крихкості при цитогенетичному аналізі. Вони, однак, передають Х-хромосому своїм дочкам, к-які можуть передавати її своїм синам. Ще однією складністю яв-ся те, що при повторних цитогенетичних обстеженнях крихкий ділянку не виявляється більш ніж у 50% жінок-носіїв. історичний екскурс З початку XX в. дослідники помітили в деяких сім'ях підвищений відсоток (часто близько 25%) осіб із затримкою психічного розвитку. Мартін і Белл в 1943 р, а ін. Пізніше, описали сім'ї, в яких брало затримка псіхіч. розвитку передавалася в формі, зчепленої з Х-хромосомою. Хоча Лабс вперше описав крихкий ділянку X в 1969 р, його опис привернуло мало інтересу, поки Сазерленд не повідомила про те, що крихкість проявляється лише при вирощуванні лімфоцитів в культурному середовищі з дефіцитом фолієвої кислоти. Відкриття чутливості fra (X) і деяких ін. Тендітних локалізацій до фолієвої кислоти дозволило підвищити точність діагностики, що, в свою чергу, призвело до виявлення великої поширеності fra (X) і зростаючому інтересу до пов'язаної з Х-хромосомою затримки псіхіч. розвитку. Наступні дослідні. підтвердили висновки Мартіна і Белла про роль fra (X) в генезі розумової відсталості. Характеристики Близько 66% дорослих пацієнтів чоловічої статі демонструють «клінічну тріаду»: а) помірну до важкої ступінь затримки розумового розвитку, б) характерні риси черепа і особи, що включають високий лоб, виступаючий підборіддя і подовжені вуха; в) збільшені тестікули (макроорхідізм). Однак індивідуальна варіабільность ознак настільки висока, що впевнена постановка діагнозу можлива лише при цитогенетичному аналізі. Розкид ознак ще вище у жінок і осіб чоловічої статі в препубертатном періоді. Хоча більшість пацієнтів чоловічої статі демонструє «синдром випередження зростання» з народження (розміри голови, джерельця і \u200b\u200bтіла перевищують 97-й процентиль), макроорхідізм і краніофаціальної риси у хлопчиків в препубертатном періоді виражені значно менш чітко. Наведені далі ознаки патології осн. на наступних публікаціях: Вregтап, Dykens, Watson, Ort, and Leckman; Brown et al .; Curfs et al .; Dykens & Leckman; Fryns; Hagerman. Фізичні ознаки. Крім характерних рис черепа і особи, макроорхідізма, випередження зростання, у осіб чоловічої статі з fra (X) можуть спостерігатися і такі ознаки, як підвищена рухливість в суглобах, високий звід неба, пролапс мітрального клапана (з шумом при аускультації), плоскостопість і низький м'язовий тонус. Жіночі носії, особливо що знаходяться на нижній межі норми розвитку інтелекту, тж демонструють такі ознаки, як високий і широкий лоб, довге обличчя і підвищена рухливість в суглобах. Когнітивні ознаки. Приблизно у 70% пацієнтів чоловічої статі є затримка розумового розвитку в помірній та тяжкій ступеня, але в цілому розмах коливань - від легкої до важкого ступеня; невеликий відсоток випадків має інтелект на рівні нижньої межі норми. Зниження рівня інтелекту у чоловіків з віком виявляється не тотальним. Порушення тут стосуються, перш за все, послідовної обробки інформ., Зниження короткочасної пам'яті на серійно пропоновану інформ., Імітації послідовних поведінкових актів. Цей дефіцит в послідовній обробці інформ. відрізняє осіб з fra (X) від пацієнтів з ін. формами розумової відсталості. Хворі чоловічої статі відносно добре справляються з завданнями, які вимагають одночасної (симультанній) обробки інформ. і інтеграції (напр., складання кубиків). Хоча більшість носіїв fra (X) має інтелект в межах норми, близько 30% хворих жіночої статі виявляють затримку розумового розвитку. Навіть ті, хто мають середній рівень інтелекту, досить часто демонструють недостатню специфічну здатність до навчання. У пацієнток тж виявляються порушення послідовної обробки інформ. і дефіцит візуально-просторових навичок, вони погано виконують цифрові і арифметичні тести, завдання з кубиками. Є недо-які дані про те, що рівень IQ у хворих жіночої статі, на відміну від чоловіків, з віком підвищується. Мова. У хворих чоловічої статі мовні навички розвиваються з відставанням. Крім того, часто спостерігаються специфічні проблеми - персевераціі, ехолалія, порушення швидкості мови, - частина яких брало може бути наслідком загальної дефицитарности послідовної обробки інформ. Поведінкові ознаки. Гіперактивність і дефіцит уваги яв-ся особливо частими наслідками fra (X), особливо у осіб чоловічої статі. У мн. часто спостерігається тенденція до соц. ізоляції, уникнення контакту поглядів і самоповреждающему поведінку, особливо нанесення собі укусів. Ставлення до інших розладів. У пацієнтів чоловічої статі може тж відзначатися судомний синдром. Fra (X) має тж коморбидность з аутизмом. У мн. пацієнтів чоловічої статі з fra (X) виявляються ознаки аутизму, в той же час у мн. хворих чоловічої статі з діагнозом аутизму виявляється fra (X) на цитогенетичному тесті. Варіабільность збігів в різних роботах досить висока, ймовірно, внаслідок розбіжності використання діагностичних критеріїв аутизму. Проте рекомендація проводити всім пацієнтам чоловічої статі з діагнозом аутизму скринінг на fra (X) видається виправданою. Терапія фолієвою кислотою Фолієва кислота часто використовувалася в спробах зниження симптоматики, зумовленої fra (X). Исслед. подвійним сліпим методом показують, що це лікування не позначається на показниках тестів інтелекту, але може знизити гіперактивність і поліпшити зосередження уваги. Питання про те, яв-ся чи лікування фолієвою кислотою настільки ж ефективним, як терапія стимуляторами, традиційно застосовуються при гиперактивном розладі з дефіцитом уваги (ГРДВ), залишається відкритим. Психол. підхід яв-ся важливим в исслед. fra (X), щонайменше, з двох причин: а) диффузность і варіабільность фіз. змін при fra (X) надає додаткове значення ролі психол. обстеження для виявлення дітей, що мають показання до цитогенетичного аналізу; б) когнітивні характеристики пацієнтів з fra (X) важливі для побудови лікувальних і пед. програм. Різноманітність проблем, пов'язаних c fra (X), роблять виправданою рекомендацію використання бригадного підходу в лікуванні таких хворих. Далі, чітке розходження між ознаками синдрому fra (X) і синдрому Дауна означає гетерогенність цих груп органічного зниження інтелекту, що має теорет. значимість. Синдром fra (X) може бути ідентифікований до народження спеціальним тестом, але неповна пенетрантність у чоловіків, фенотипічні ефекти у значної кількості носіїв жіночої статі і труднощі діагностики у осіб жіночої статі з невираженою симптоматикою ускладнюють генетичне консультування. Нарешті, хоча багато відомо про fra (X), зауваження Уеллс і Браун про те, що «наступні дослідні. можуть змінити суч. подання щодо тендітної Х-хромосоми », представляється розумним застереженням.
Це одна з найбільш часто зустрічаються форм розумової відсталості після хвороби Дауна. Популяційна частота 1: 2000 - 1: 2500 новонароджених. Хворих хлопчиків в 2-3 рази більше, ніж дівчаток і хворіють вони важче. Зовнішність хворих не завжди специфічна. Характерно довгасте обличчя, високий виступаючий лоб, макро- і долихоцефалия, гіпоплазія середньої частини обличчя. Губи товсті, нижня губа часто вивернута. Характерна макроотія, великі кисті і стопи. Типовий симптом - макроорхідізм - з'являється у підлітків. Яєчка збільшені за рахунок розвитку сполучної тканини. Статева активність мінімальна. Спостерігаються симптоми вродженої дисплазії сполучної тканини - слабкість зв'язок суглобів, плоскостопість, іноді деформація хребта і ін. Розумова відсталість частіше помірна, рідко глибока (10-15%). Більшість хворих соціально адаптовані, можуть виконувати нескладну фізичну роботу. У жінок носії знижений IQ. Захворювання обумовлене мутацією гена FMR-1 (fragile mental retardation), який локалізується в довгому плечі Х-хромосои (Xq). Патологічний ген має велику кількість тринуклеотидних повторів (РГД) в 5 1 - нетрансльовані області цього гена. У нормі кількість повторів від 6 до 42. Хромосома з 50-200 повторами вважається премутаціей. У наступному поколінні число повторів може збільшуватися до 1000, що і призводить до вираженої клінічної картині. Клінічна картина залежить від числа повторів. Якщо жінка успадкувала велику кількість повторів, то вона теж буде боліти. Але це зустрічається рідко. Основний метод діагностики - кариотипирование. Лімфоцити хворого культивують в середовищі без фолієвої кислоти. У довгому плечі Х-хромосоми виявляють "ламкий" ( "фрагільної ділянку"). Зовні нагадує вторинну перетяжку і супутник. Хромосома пошкоджується в області великого числа тринуклеотидних повторів. Можлива молекулярно-генетична діагностика, розроблені методи пренатальної діагностики. Фосфат-діабет (гіпофосфатемія)Тип успадкування Х-зчеплений домінантний. Ген локалізованість Хр 22.2 - р 22.І. Гіпофосфатемію можна виявити відразу після народження, а ознаки рахіту з'являються в кінці першого - початку другого року життя, коли діти починають ходити. Більше виражені зміни нижніх кінцівок: варусне викривлення довгих трубчастих кісток. Характерні низький зріст, обмеження рухливості в великих суглобах (тазостегнових, колінних, ліктьових), долихоцефалия, дисплазія нігтів. Хода невпевнена, в важких випадках хворі взагалі не можуть ходити. На відміну від вітамін - Д - дефіцитного рахіту загальний стан не порушено. У жінок скелетні порушення менш виражені. Рентгенологічно виявляються типові для рахіту зміни - грубоволокниста структура губчастої речовини кісток. У крові підвищений рівень лужної фосфатази, рівень кальцію в нормі. Захворювання обумовлене зниженням реабсорбції фосфатів у канальцях нирок. Спадкові захворювання зчеплені з Х-хромосомою. |
Назва захворювання або синдрому.
|
Популяційна частота.
|
Локалізація гена.
|
Мінімальні діагностичні ознаки.
|
|
Х-зчеплені рецесивні
|
|
Гемофілія А порушення синтезу VIII фактора згортання крові
|
1: 2500 хлопчиків
|
|
Тривалі кровотечі при травмах, гемартрози (крововиливи у великі суглоби - колінний, ліктьовий, гомілковостопний), знижена прокоагулянтна активність фактора VIII. Подовження часу згортання крові.
|
|
дальтонізм
|
|
|
Нерозрізнення червоного і зеленого кольорів.
|
|
Леша- Нихана синдром.
|
|
|
Описано в порушеннях обміну речовин.
|
|
М'язова дистрофія Дюшенна -Беккера (Псевдогіпертрофічна). Порушення синтезу білка дистрофина сарколемми клітин скелетної мускулатури. Загибель скелетної мускулатури і заміна її жирової та сполучної тканиною
|
Дюшенна- 1: 3000 хлопчиків Беккера-1: 30000 хлопчиків
|
|
М'язова слабкість переважно в проксимальних групах м'язів, псевдогіпертрофія м'язів (литкових, сідничних, дельтовидних і т.д.), підвищення рівня креатинфосфокінази в сироватці крові. У 10-15 років хворі прикуті до ліжка, в 20-30 вмирають. Форма Беккера - більш доброякісний перебіг.
|
|
Синдром Кріста-Симменса-Турена (ангідротіческая ектодермальна дисплазія).
|
невідома
|
|
Гіпогідроз (гіпоплазія потових залоз), порушення терморегуляції. Гіподонтія, шиловидні зуби. Гіпотрихоз. Суха шкіра і слизові.
|
|
Домінантне зчеплене з Х-хромосомою спадкування
|
|
Фосфат-діабет (вітамін Д резистентний рахіт) - порушення реабсорбції фосфатів у канальцях нирок.
|
|
Xp 22.2- p 21.2
|
Рахіт, що не піддається лікуванню вітаміном Д. Симптоми рахіту з'являються в кінці 1 - на 2 році життя. Характерна варусна деформація нижніх кінцівок. Гипофосфатемия. Підвищено рівень лужної фосфатази в крові, рівень кальцію в нормі.
|
Керівник напрямку
"Онкогенетики"
Жусіна
Юлія Геннадіївна
Закінчила педіатричний факультет Воронезького державного медичного університету ім. М.М. Бурденко в 2014 році.
2015 - інтернатура по терапії на базі кафедри факультетської терапії ВГМУ ім. М.М. Бурденко.
2015 - сертифікаційний курс за фахом «Гематологія» на базі гематологічного наукового центру м Москви.
2015-2016 - лікар терапевт ВГКБСМП №1.
2016 - затверджена тема дисертації на здобуття наукового ступеня кандидата медичних наук «вивчення клінічного перебігу захворювання та прогнозу у хворих хронічними обструктивними захворюваннями легень з анемічним синдромом». Співавтор понад 10 друкованих праць. Учасник науково-практичних конференцій з генетики і онкології.
2017 - курс підвищення кваліфікації за темою: «інтерпретація результатів генетичних досліджень у хворих зі спадковими захворюваннями».
З 2017 року ординатура за спеціальністю «Генетика» на базі РМАНПО.
Керівник напрямку
"Генетика"
Канівець
Ілля Вячеславович

Канівець Ілля Вячеславович, лікар-генетик, кандидат медичних наук, керівник відділу генетики медико-генетичного центру Геномед. Асистент кафедри медичної генетики Російської медичної академії неперервної професійної освіти.
Закінчив лікувальний факультет Московського державного медико-стоматологічного університету в 2009 році, а в 2011 - ординатуру за фахом «Генетика» на кафедрі Медичної генетики того ж університету. У 2017 році захистив дисертацію на здобуття наукового ступеня кандидата медичних наук на тему: Молекулярна діагностика варіацій числа копій ділянок ДНК (CNVs) у дітей з вродженими вадами розвитку, аномаліями фенотипу і / або розумовою відсталістю при використанні SNP олігонуклеотидних микроматриц високої щільності »
C 2011-2017 працював лікарем-генетиком в Дитячій клінічній лікарні ім. Н.Ф. Філатова, науково-консультативному відділі ФГБНУ «Медико-генетичний науковий центр». З 2014 року по теперішній час керує відділом генетики МГЦ Геномед.
Основні напрямки діяльності: діагностика та ведення пацієнтів зі спадковими захворюваннями та вродженими вадами розвитку, епілепсією, медико-генетичне консультування сімей, в яких народилася дитина зі спадковою патологією або вадами розвитку, пренатальна діагностика. У процесі консультації проводиться аналіз клінічних даних і генеалогії для визначення клінічної гіпотези і необхідного обсягу генетичного тестування. За результатами обстеження проводиться інтерпретація даних і роз'яснення отриманої інформації консультирующимся.
Є одним із засновників проекту «Школа Генетики». Регулярно виступає з доповідями на конференціях. Читає лекції для лікарів генетиків, неврологів і акушерів-гінекологів, а також для батьків пацієнтів зі спадковими захворюваннями. Є автором і співавтором понад 20 статей і оглядів в російських і зарубіжних журналах.
Область професійних інтересів - впровадження сучасних повногеномне досліджень в клінічну практику, інтерпретація їх результатів.
Час прийому: СР, ПТ 16-19
Керівник напрямку
"Неврологія"
Шарков
Артем Олексійович

Шарков Артем Олексійович - лікар-невролог, епілептології
У 2012 році навчався за міжнародною програмою "Oriental medicine" в університеті Daegu Haanu в Південній Кореї.
З 2012 року - участь в організації бази даних і алгоритму для інтерпретації генетичних тестів xGenCloud (https://www.xgencloud.com/, Керівник проекту - Ігор Угаров)
У 2013 році закінчив педіатричний факультет Російського національного дослідницького медичного університету імені М.І. Пирогова.
C 2013 по 2015 рік навчався в клінічній ординатурі з неврології в ФГБНУ «Науковий центр неврології».
З 2015 року працює неврологом, науковим співробітником в Науково дослідному клінічному інституті педіатрії імені академіка Ю. Є. Вельтіщева ГБОУ ВПО РНІМУ ім. Н.І. Пирогова. Також працює лікарем-неврологом і лікарем лабораторії відео-ЕЕГ моніторингу в клініках «Центр епілептології та неврології ім. А.А.Казаряна »і« епілепсія-центр ».
У 2015 році пройшов навчання в Італії на школі «2nd International Residential Course on Drug Resistant Epilepsies, ILAE,-2015».
У 2015 році підвищення кваліфікації - «Клінічна і молекулярна генетика для практикуючих лікарів», РДКБ, РОСНАНО.
У 2016 році підвищення кваліфікації - «Основи молекулярної генетики» під керівництвом біоінформатика, к.б.н. Коновалова Ф.А.
З 2016 року - керівник неврологічного напрямки лабораторії "Геномед".
У 2016 році пройшов навчання в Італії на школі «San Servolo international advanced course: Brain Exploration and Epilepsy Surger, ILAE, 2016».
У 2016 році підвищення кваліфікації - "Інноваційні генетичні технології для лікарів", "Інститут лабораторної медицини".
У 2017 році - школа «NGS в медичній генетиці 2017», МГНЦ
В даний час проводить наукові дослідження в галузі генетики епілепсії під керівництвом професора, д.м.н. Білоусової Е.Д. і професора, д.м.н. Дада Е.Л.
Затверджена тема дисертації на здобуття наукового ступеня кандидата медичних наук "Клініко-генетичні характеристики моногенних варіантів ранніх епілептичних енцефалопатій".
Основні напрямки діяльності - діагностика і лікування епілепсії у дітей та дорослих. Вузька спеціалізація - хірургічне лікування епілепсії, генетика епілепсій. Нейрогенетика.
Научні публікації
Шарков А., Шаркова І., Головтеев А., Угаров І. «Оптимізація диференціальної діагностики та інтерпретації результатів генетичного тестування експертної системою XGenCloud при деяких формах епілепсій». Медична генетика, № 4, 2015 року, с. 41.
*
Шарков А.А., Воробйов А.Н., Троїцький А.А., Савкіна І.С., Дорофєєва М.Ю., Мелікян А.Г., Головтеев А.Л. "Хірургія епілепсії при многоочаговое ураження головного мозку у дітей з туберозний склерозом." Тези XIV Російського Конгресу «ІННОВАЦІЙНІ ТЕХНОЛОГІЇ В ПЕДІАТРІЇ І ДИТЯЧОЇ ХІРУРГІЇ». Російський Вісник перинатології та педіатрії, 4, 2015. - с.226-227.
*
Дада Е.Л., Білоусова Е.Д., Шарков А.А. "Молекулярно-генетичні підходи до діагностики моногенних идиопатических і симптоматичних епілепсій". Теза XIV Російського Конгресу «ІННОВАЦІЙНІ ТЕХНОЛОГІЇ В ПЕДІАТРІЇ І ДИТЯЧОЇ ХІРУРГІЇ». Російський Вісник перинатології та педіатрії, 4, 2015. - с.221.
*
Шарков А.А., Дада Е.Л., Шаркова І.В. «Рідкісний варіант ранньої епілептичної енцефалопатії 2 типу, зумовленої мутаціями в гені CDKL5 у хворого чоловічої статі». Конференція "епілептології в системі нейронаук". Збірник матеріалів конференції: / За редакцією: проф. Незнанова Н.Г., проф. Михайлова В.А. СПб .: 2015. - с. 210-212.
*
Дада Е.Л., Шарков А.А., Канівець І.В., Гундорова П., Фоміних В.В., Шаркова І, В ,. Троїцький А.А., Головтеев А.Л., Поляков А.В. Новий алельних варіант миоклонус-епілепсії 3 типу, обумовлений мутаціями в гені KCTD7 // Медична генетіка.-2015.- т.14.-№9.- С.44-47
*
Дада Е.Л., Шаркова І.В., Шарков А.А., Акімова І.А. «Клініко-генетичні особливості і сучасні способи діагностики спадкових епілепсій». Збірник матеріалів «Молекулярно-біологічні технології в медичній практиці» / Под ред. чл.-кор. РАПН А.Б. Масленнікова.- Вип. 24.- Новосибірськ: Академіздат, 2016.- 262: с. 52-63
*
Білоусова Е.Д., Дорофєєва М.Ю., Шарков А.А. Епілепсія при туберозному склерозі. В "Хвороби мозку, медичні та соціальні аспекти" під редакцією Гусєва Є.І., Гехт А.Б., Москва; 2016 року; стр.391-399
*
Дада Е.Л., Шарков А.А., Шаркова І.В., Канівець І.В., Коновалов Ф.А., Акімова І.А. Спадкові захворювання і синдроми, що супроводжуються фебрильними судомами: клініко-генетичні характеристики і способи діагностики. // Російський Журнал Дитячої Неврологіі.- Т. 11.- №2, с. 33- 41. doi: 10.17650 / 2073-8803- 2016-11- 2-33- 41
*
Шарков А.А., Коновалов Ф.А., Шаркова І.В., Білоусова Е.Д., Дада Е.Л. Молекулярно-генетичні підходи до діагностики епілептичних енцефалопатій. Збірник тез «VI БАЛТІЙСЬКИЙ КОНГРЕС З ДИТЯЧОЇ НЕВРОЛОГІЇ» / За редакцією професора Гузева В.І. Санкт-Петербург, 2016, с. 391
*
Гемісферотоміі при фармакорезистентній епілепсії у дітей з білатеральним ураженням головного мозку Зубкова Н.С., Алтунина Г.Є., Землянський М.Ю., Троїцький А.А., Шарков А.А., Головтеев А.Л. Збірник тез «VI БАЛТІЙСЬКИЙ КОНГРЕС З ДИТЯЧОЇ НЕВРОЛОГІЇ» / За редакцією професора Гузева В.І. Санкт-Петербург, 2016, с. 157.
*
*
Стаття: Генетика і диференційоване лікування ранніх епілептичних енцефалопатій. А.А. Шарков *, І.В. Шаркова, Е.Д. Білоусова, Е.Л. Дада. Журнал неврології і психіатрії, 9, 2016 року; Вип. 2doi: 10.17116 / jnevro 20161169267-73
*
Головтеев А.Л., Шарков А.А., Троїцький А.А., Алтунина Г.Є., Землянський М.Ю., Копачів Д.Н., Дорофєєва М.Ю. "Хірургічне лікування епілепсії при туберозному склерозі" під редакцією Дорофеєвої М.Ю., Москва; 2017; стр.274
*
Нові міжнародні класифікації епілепсій та епілептичних нападів Міжнародної Ліги по боротьбі з епілепсією. Журнал неврології і психіатрії ім. C.C. Корсакова. 2017. Т. 117. № 7. С. 99-106
Керівник напрямку
"Пренатальна діагностика"
Київська
Юлія Кирилівна

У 2011 році закінчила Московський Державний Медико-Стоматологічний Університет ім. А.І. Євдокимова за фахом «Лікувальна справа» Навчалася в ординатурі на кафедрі Медичної генетики того ж університету за спеціальністю «Генетика»
У 2015 році закінчила інтернатуру за фахом Акушерство і Гінекологія в Медичному інституті удосконалення лікарів ФГБОУ ВПО «МГУПП»
З 2013 року веде консультативний прийом в ГБУЗ «Центр Планування Сім'ї і Репродукції» ДЗМ
З 2017 року є керівником напряму «Пренатальна діагностика» лабораторії Геномед
Регулярно виступає з доповідями на конференціях і семінарах. Читає лекції для лікарів різних спеціальної в області репродуціі і пренатальної діагностики
Проводить медико-генетичне консультування вагітних з питань пренатальної діагностики з метою попередження народження дітей з вродженими вадами розвитку, а так само сімей з імовірно спадкової або вродженою патологією. Проводить інтерпретацію отриманих результатів ДНК-діагностики.
ФАХІВЦІ
Латипов
Артур Шамилевич

Латипов Артур Шамилевич - лікар генетик вищої кваліфікаційної категорії.
Після закінчення в 1976 році лікувального факультету Казанського державного медичного інституту протягом багатьох працював спочатку лікарем кабінету медичної генетики, потім завідувачем медико-генетичним центром Республіканської лікарні Татарстану, головним фахівцем міністерства охорони здоров'я Республіки Татарстан, викладачем кафедр Казанського медуніверситету.
Автор понад 20 наукових праць з проблем репродукционной і біохімічної генетики, учасник багатьох вітчизняних і міжнародних з'їздів і конференцій з проблем медичної генетики. Впровадив в практичну роботу центру методи масового скринінгу вагітних і новонароджених на спадкові захворювання, провів тисячі інвазивних процедур при підозрі на спадкові захворювання плода на різних термінах вагітності.
З 2012 року працює на кафедрі медичної генетики з курсом пренатальної діагностики Російської академії післядипломної освіти.
Область наукових інтересів - метаболічні хвороби у дітей, допологова діагностика.
Час прийому: СР 12-15, СБ 10-14
Прийом лікарів здійснюється за попереднім записом.
Лікар-генетик
Габелко
Денис Ігорович

У 2009 році закінчив лікувальний факультет КДМУ ім. С. В. Курашова (спеціальність «Лікувальна справа»).
Інтернатура в Санкт-Петербурзької медичної академії післядипломної освіти Федерального агентства з охорони здоров'я і соціального розвитку (спеціальність «Генетика»).
Інтернатура по терапії. Первинна перепідготовка за спеціальністю «Ультразвукова діагностика». З 2016 року є співробітником кафедри кафедри фундаментальних основ клінічної медицини інституту фундаментальної медицини та біології.
Сфера професійних інтересів: пренатальна діагностика, застосування сучасних скринінгових та діагностичних методів для виявлення генетичної патології плоду. Визначення ризику повторного виникнення спадкових хвороб в сім'ї.
Учасник науково-практичних конференцій з генетики та акушерства і гінекології.
Стаж роботи 5 років.
Консультація за попереднім записом
Прийом лікарів здійснюється за попереднім записом.
Лікар-генетик
Гришина
Христина Олександрівна

Закінчила в 2015 році Московський Державний Медико-Стоматологічний Університет за спеціальністю «Лікувальна справа». У тому ж році вступила в ординатуру за фахом 30.08.30 «Генетика» в ФГБНУ «Медико-генетичний науковий центр».
Прийнята на роботу в лабораторію молекулярної генетики складно успадкованих захворювань (завідувач - д.б.н. Карпухін А.В.) в березні 2015 року на посаду лаборанта-дослідника. З вересня 2015 року переведена на посаду наукового співробітника. Є автором і співавтором понад 10 статей і тез з клінічної генетики, онкогенетики і молекулярної онкології в російських і зарубіжних журналах. Постійний учасник конференцій з медичної генетики.
Область науково-практичних інтересів: медико-генетичне консультування хворих зі спадковою синдромальной і мультифакторіальної патологією.
Консультація лікаря-генетика дозволяє відповісти на питання:
чи є симптоми у дитини ознаками спадкового захворювання
-яке дослідження необхідно для виявлення причини
визначення точного прогнозу
рекомендації по проведенню і оцінка результатів пренатальної діагностики
все, що потрібно знати при плануванні сім'ї
консультація при плануванні ЕКО
виїзні та онлайн консультації
брали участь в науково-практичній школі "Інноваційні генетичні технології для лікарів: застосування в клінічній практиці", конференції Європейського товариства генетики людини (ESHG) та інших конференціях, присвячених генетиці людини.
Проводить медико-генетичне консультування сімей з імовірно спадкової або вродженою патологією, включаючи моногенні захворювання і хромосомні аномалії, визначає показання до проведення лабораторних генетичних досліджень, проводить інтерпретацію отриманих результатів ДНК-діагностики. Консультує вагітних з питань пренатальної діагностики з метою попередження народження дітей з вродженими вадами розвитку.
Лікар-генетик, лікар акушер-гінеколог, кандидат медичних наук
Кудрявцева
Олена Володимирівна

Лікар-генетик, лікар акушер-гінеколог, кандидат медичних наук.
Спеціаліст в області репродуктивного консультування і спадкової патології.
Закінчила Уральську державну медичну академію в 2005 році.
Ординатура за спеціальністю «Акушерство і гінекологія»
Інтернатура за фахом «Генетика»
Професійна перепідготовка за спеціальністю «Ультразвукова діагностика»
Напрямки діяльності:
- Безпліддя і невиношування вагітності
Василиса Юріївна 
Є випускницею Нижегородської державної медичної академії, лікувального факультету (спеціальність «Лікувальна справа»). Закінчила клінічну ординатуру ФБГНУ «МГНЦ» за спеціальністю «Генетика». У 2014 році проходила стажування в клініці материнства і дитинства (IRCCS materno infantile Burlo Garofolo, Trieste, Italy).
З 2016 року працює на посаді лікаря-консультанта в ТОВ «Геномед».
Регулярно бере участь в науково-практичних конференціях з генетики.
Основні напрямки діяльності: Консультування з питань клінічної і лабораторної діагностики генетичних захворювань і інтерпретація результатів. Ведення пацієнтів і їх сімей з імовірно спадковою патологією. Консультування при плануванні вагітності, а також при вагітності з питань пренатальної діагностики з метою попередження народження дітей з вродженою патологією.
Q99.2 ломки Х-хромосома
Епідеміологія
Синдром Мартіна-Белла досить часте захворювання: на 1000 чоловіків припадає 0,3-1,0 страждають на цю недугу, а на 1000 жінок - 0,2-0,6. Причому діти з синдромом Мартіна-Белла народжуються на всіх континентах з однаковою періодичністю. Очевидно, національність, колір шкіри, розріз очей, умови проживання, добробут людей не впливають на виникнення недуги. Частота його виникнення порівнянна хіба що з частотою синдрому Дауна (на 600-800 новонароджених 1 захворювання). П'ята частина чоловіків-носіїв зміненого гена здорові, не мають клінічних і генних відхилень, інші з ознаки розумової відсталості від легкої до важких форм. Серед жінок-носіїв хворих трохи більше третини.
Синдром ламкої Х хромосоми зачіпає приблизно 1 на 2500-4000 чоловіків і 1 на 7000-8000 жінок. Поширеність носіїв захворювання серед жіночого носеленія, за оцінками, може досягати 1 на 130-250 чоловік; поширеність носіїв серед чоловічої статі, за оцінками, 1 на 250-800.
Причини синдрому Мартіна-Бела
Синдром Мартіна-Белла розвивається через повну або часткового припинення вироблення організмом специфічного білка. Відбувається це через відсутність реакції від гена типу FMR1, що локалізується в Х-хромосомі. Мутація виникає в результаті перебудови структури гена з нестабільних структурних варіантів станів гена (алелей), а не з самого початку. Захворювання передається тільки по чоловічій лінії, при чому чоловік не обов'язково може бути хворий. Чоловіки-носії, передають ген своїм дочкам в незміненому вигляді, тому у них розумова відсталість не очевидна. При подальшій передачі гена від матері її дітям ген мутує, тут і проявляються всі ознаки, властиві цьому захворюванню.
Фактори ризику
патогенез
В основі патогенезу синдрому Мартіна-Бела лежать мутації генного апарату, які призводять до блокування вироблення FMR-протеїну, білка, життєво необхідного організму, особливо в нейронах, і присутній у різних тканинах. Дослідження показують, що FMR-протеїни беруть безпосередню участь в процесах регуляції трансляцій, які відбуваються в тканинах мозку. Відсутність цього білка або обмежена його вироблення організмом і призводить до розумової відсталості.
У патогенезі захворювання ключовим порушенням вважається гіперметілірованіе гена, але остаточно виявити механізм розвитку даного розладу поки не вдалося.
Разом з цим також виявлена \u200b\u200bлокусная гетерогенність патології, яка пов'язана з поліаллелізмом, а також полілокусностью. Визначено наявність алельних варіантів розвитку хвороби, які обумовлюються існуванням точкових мутацій, а також знищенням гена типу FMRL.
Також у пацієнтів виявляють 2 чутливих до фолієвої кислоти фрагільної триплетів, розташованих в З00 т.п.н., а також 1,5-2 млн.п.н. від фрагільної триплета, в якому міститься ген FMR1. Механізм відбуваються в генах FRAXE, а також FRAXF (вони ідентифіковані в вищевказаних фрагільної триплетах) мутацій співвідноситься з механізмом порушень при синдромі Мартіна Белла. Обумовлюється даний механізм поширенням GCC-, а також CGG-повторів, при яких відбувається метилування так званих CpG-острівців. Крім класичної форми патології існує також 2 рідкісних типу, які відрізняються внаслідок експансії тринуклеотидних повторів (в чоловічому і в жіночому мейозе).
Виявлено, що при класичній формі синдрому у хворого відсутній особливий нуклеоцітоплазматіческій білок типу FMR1, який виконує функцію зв'язування різноманітних мРНК. Крім цього даний білок сприяє утворенню комплексу, який допомагає здійснювати трансляційні процеси всередині рибосом.
Симптоми синдрому Мартіна-Бела
Як розпізнати захворювання у дітей? Які перші ознаки? У перші місяці життя дитини розпізнати симптом Мартіна-Белла можна, хіба що іноді спостерігається зниження м'язового тонусу. Після року клініка захворювання більш очевидна: дитина починає пізно ходити і говорити, іноді мова повністю відсутня. Він гіперактивна, безладно розмахує руками, боїться натовпу і шуму, упертий, спостерігаються різкі спалахи гніву, емоційна нестабільність, трапляються епілептичні припадки, не йде на очної контакт. У хворих синдромом Мартіна-Белла захворювання видає і зовнішність: вуха, відстовбурчені і великі, лоб важкий, обличчя видовжене, підборіддя виступає, косоокість, широкі кисті і стопи. Їм властиві і ендокринні порушення: часто велику вагу, ожиріння, у чоловіків великі яєчка, раннє статеве дозрівання.

Серед хворих синдромом Мартіна-Белла рівень інтелекту сильно відрізняється: від невеликої розумової відсталості до її важких випадків. Якщо у нормальної людини коефіцієнт інтелекту (IQ) у середньому дорівнює 100, а у генія - 130, то у людей, схильних до недугу - 35-70.
Всі клінічні симптоми патології можна охарактеризувати тріадою основних проявів:
- олігофренія (показник IQ дорівнює 35-50);
- дисморфофобія (спостерігаються відстовбурчені вуха, а також прогнатизм);
- макроорхідізм, який проявляється після настання статевої зрілості.
Приблизно у 80% хворих виявляють також пролапс двостулкового клапана.
Але при цьому повна форма синдрому проявляється лише у 60% всіх хворих. У 10% виявляють лише розумову відсталість, а у інших захворювання розвивається з різною комбінацією ознак.
Серед перших ознак хвороби, що проявляються вже в ранньому віці:
- у хворої дитини спостерігається значна розумова відсталість в порівнянні з розвитком інших однолітків;
- розлади уваги і зосередженості;
- сильне впертість;
- діти досить пізно починають ходити і розмовляти;
- спостерігаються гіперактивність і порушення в розвитку мови;
- дуже сильні і неконтрольовані напади гніву;
- може розвиватися мутизм - це повна відсутність у дитини мовлення;
- малюк відчуває соціальне занепокоєння, здатний панікувати через гучного шуму або будь-яких інших сильних звуків;
- дитина неконтрольовано і хаотично розмахує руками;
- спостерігається боязкість, дитина боїться перебування в місцях великого скупчення народу;
- виникнення різних нав'язливих ідей, нестабільне емоційний стан;
- у малюка може спостерігатися небажання встановлювати зоровий контакт з людьми.
У дорослих спостерігаються такі симптоми патології:
- специфічна зовнішність: видовжене обличчя з важким чолом, великі відстовбурчені вуха, сильно виступаючий вперед підборіддя;
- плоскостопість, отит і косоокість;
- статеве дозрівання наступає досить рано;
- може розвиватися ожиріння;
- досить часто при синдромі Мартіна Белла спостерігаються пороки в розвитку серця;
- у чоловіків спостерігається збільшення яєчок;
- зчленування суглобів стають дуже рухливими;
- різко збільшується вага, а також зростання.
Діагностика синдрому Мартіна-Бела
Для діагностики синдрому Мартіна Белла потрібно звертатися до кваліфікованого генетику. Діагноз ставиться після проведення специфічних генетичних тестів, які дозволяють визначити дефектну хромосому.
аналізи
На ранній стадії розвитку захворювання використовується цитогенетичний метод, при якому у пацієнта беруть фрагмент клітинного матеріалу, до якого потім додають фолієву кислоту, щоб спровокувати в хромосомах зміни. Після деякого періоду часу виявляється область хромосоми, на якій спостерігається помітне стоншення - це і є ознакою наявності синдрому фрагільної Х-хромосоми.
Але даний аналіз не підходить для діагностики на пізніх етапах хвороби, тому як його точність знижується внаслідок широкого застосування полівітамінів, що містять фолієву кислоту.
Інтегрованої діагностикою синдрому Мартіна-Белла є молекулярно-генетична експертиза, яка полягає у визначенні кількості так званих тринуклеотидних повторів в гені.
інструментальна діагностика
Високоспецифічний методом інструментальної діагностики є ПЛР (полімеразна ланцюгова реакція), що дозволяє вивчити структуру містяться в Х-хромосомі залишків амінокислот і тим самим визначити наявність синдрому Мартіна Белла.
Існує також окремий, навіть більш специфічний, метод діагностики патології - поєднання ПЛР і детекції за допомогою капілярного електрофорезу. Даний метод є високоточним і виявляє хромосомну патологію у хворих, що мають первинну форму яєчникової недостатності, а також атаксические синдром.
Визначити наявність дефекту можна після здійснення діагностики на ЕЕГ. У пацієнтів з цим захворюванням відзначається схожа біоелектрична мозкова активність.
Диференціальна діагностика
До диференційованим методам, що допомагає запідозрити синдром відносяться:
- клінічний - 97,5% хворих мають очевидні ознаки розумової відсталості (помірною або глибокою); у 62% - відстовбурчені великі вуха; у 68,4% - великий видатний вперед підборіддя і лоб; у 68,4% хлопчиків - збільшені яєчка, у 41,4% - особливості мови (темп мови нерівномірний, гучність некерована і ін.);
- цитогенических - досліджуються кров на культуру лімфоцитів, визначають кількість клітин з ламкою Х-хромосомою на 100 вивчених клітин;
- електроенцефалографія - реєструються специфічні для синдрому Мартіна-Бела зміни електричних імпульсів роботи головного мозку.
Лікування синдрому Мартіна-Бела
При лікуванні дорослих пацієнтів застосовуються антидепресанти з психостимуляторами. Процес медикаментозної терапії постійно контролюється психологом і психіатром. Крім цього в приватних клініках виконуються процедури мікроін'єкцій такими ліками, як Церебролізин (або його деривати), а також цітомедіни (такі, як Солкосерил або Лидаза).
При розвитку атаксические синдрому застосовують ЛЗ, що розріджують кров, а також ноотропи. Крім цього призначаються амінокислотні суміші і Ангіопротектори. Жінкам з первинною формою недостатності яєчників призначають коригуючий лікування з використанням фітолекарств і естрогенів.
Також при лікуванні застосовуються антагоністи глутамінової рецепторів.
Традиційним для лікування синдрому Мартіна-Белла є застосування медикаментозних препаратів, які впливають на симптоми захворювання, але не на його причину. Дана терапія полягає в призначенні антидепресантів, нейролептиків, психостимуляторів. Не всі препарати показані до застосування дітям, тому список лікарських засобів досить обмежений. До нейролептиків, які можна застосовувати після 3-х років (самий ранній вік їх призначення) відноситься галоперидол в краплях і таблетках, хлорпромазин в розчині, періціазін в краплях. Так, доза прийому галоперидолу для дітей розраховується залежно від маси тіла. Для дорослих доза призначається індивідуально. Приймають всередину, починають з 0,5-5 мг 2-3 рази на добу, потім дозу поступово збільшують до 10-15 мг. Коли настає поліпшення, переходять до більш низьких доз, для підтримки досягнутого стану. При психомоторному збудженні призначають 5-10мг внутрішньом'язово або внутрішньовенно, можливі кілька повторів через 30-40 хв. Добова доза не повинна перевищувати 100 мг. Можливі побічні явища у вигляді нудоти, блювоти, спазмованих м'язів, підвищення тиску, аритмії і т.д. Особливі запобіжні заходи повинні дотримуватися люди похилого віку, тому що зареєстровані випадки раптової зупинки серця, можлива поява пізньої дискінезії (виникнення мимовільних рухів).
Антидепресанти підвищують діяльність структур головного мозку, знімають пригнічений настрій, напруженість, піднімають настрій. До даних препаратів, рекомендованих до прийому з 5-8 років при синдромі Мартіна-Белла, відносяться кломіпромін, сертралін, флюоксегін, флювоксамін. Так, флюоксетин приймається під час їжі всередину 1-2 (бажано в першій половині дня), починають з 20 мг на добу, збільшуючи до 80мг при необхідності. Людям похилого віку не рекомендують дозу вище 60 мг. Курс лікування визначає лікар, але не більше 5 тижнів.
Можливі побічні реакції: запаморочення, тривожність, шум у вухах, зниження апетиту, тахікардія, набряки і т.д. Необхідно виявляти обережність при призначенні літнім людям, з серцево-судинними захворюваннями, цукровим діабетом.
Психостимулятори - психотропні препарати, застосовують з метою посилити сприйняття зовнішніх подразників: загострюють слух, відповідні реакції, зір.
Як седативного препарату при неврозах, тривожних станах, епілептичних припадках, судомах призначають діазепам. Приймається всередину, внутрішньовенно, внутрішньом'язово, ректально (в пряму кишку). Призначається індивідуально, залежно від тяжкості захворювання, з найменших доз 5-10мг, добова - 5-20мг. Тривалість лікування 2-3 місяці. Для дітей дозу розраховують з урахуванням маси тіла і індивідуальних особливостей. До побічних явищ належить млявість, апатія, сонливість, нудота, запор. Небезпечно поєднувати з алкоголем, можливо звикання до препарату.
При лікуванні синдрому Мартіна-Бела зафіксовані випадки поліпшення стану і при введенні препаратів, виготовлених на основі матеріалу тваринного походження (мозку): церебролізата, церебролізину, церебролізат-М. Основними компонентами цих препаратів є пептиди, які сприяють виробленню білка в нейронах, таким чином поповнюється відсутній білок. Церебролізин вводять струминно по 5-10мл, курс лікування складається з 20-30 ін'єкцій. Дітям препарат призначають з року життя, вводять внутрішньом'язово щодня по1-2мл протягом місяця. Можливі повторні сеанси прийому. Побічні явища у вигляді спека, протипоказаний вагітним жінкам.
Були спроби лікувати недугу фолієвою кислотою, але поліпшувався лише поведінковий аспект (знижувався рівень агресії, гіперактивності, поліпшувалася мова) а на інтелектуальному рівні нічого не змінювалося. Для поліпшення станів при захворюванні призначають фолієву кислоту, методи фізіотерапії, показана логопедична, педагогічна і соціальна корекція.
Ефективними також вважаються препарати літію, які допомагають поліпшити адаптацію хворого в соціальному середовищі, а також когнітивну діяльність. Крім цього вони ще регулюють його поведінку в суспільстві.
Застосування трав при синдромі Мартіна-Белла можливо в якості антидепресантів. До травам, що допомагає зняти напругу, тривожність, поліпшити сон відносяться валеріана, м'ята перцева, чебрець, звіробій, ромашка. Настої готують так: на 1 чайну ложку сухих трав знадобиться склянку окропу, відвари наполягають не менше 20хв, приймають переважно на ніч перед сном або ж у другій половині дня. Непоганий добавкою до них буде ложка меду.
фізіотерапевтичне лікування
Щоб усунути неврологічні прояви, проводяться спеціальні фізіотерапевтичні процедури - такі, як вправи в басейні, релаксація м'язів і голковколювання.
оперативне лікування
Важливим етапом лікування також вважаються методи пластичної хірургії - операції, що допомагають поліпшити зовнішність хворого. Проводиться пластика кінцівок і вушних раковин, а крім цього статевих органів. Виконується також корекція гинекомастии з Епіспадія, а разом з цим інших недоліків зовнішнього вигляду.
профілактика
Єдиний метод профілактики захворювання - це пренатальний скринінг вагітних. Існують спеціальні обстеження, які дозволяють ще на ранньому етапі визначити наявність патології, після чого рекомендується перервати вагітність. Як альтернативу використовують ЕКО, здатне допомогти тому, щоб дитина змогла успадкувати здорову хромосому Х.
], [
|